This page is intended for Healthcare Professionals.
PRISMS study
PRISMS study
IN PRISMS, REBIF® DEMONSTRATED EFFICACY VS PLACEBO IN RELAPSE REDUCTION, LESION REDUCTION, AND DELAYS IN DISABILITY PROGRESSION1-3
Jump to topic:
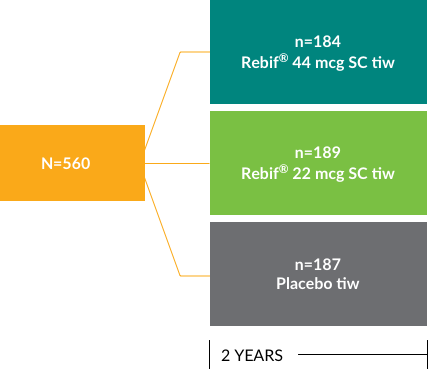
PRISMS study design
Prevention of Relapses and Disability by Interferon β-1a Subcutaneously in Multiple Sclerosis (PRISMS) was a double-blind, placebo-controlled study conducted over 2 years. Patients were randomly assigned Rebif® 44 mcg (n=184), Rebif® 22 mcg (n=189), or placebo (n=187), given 3 times weekly by subcutaneous injection. Outcomes measured included relapse rate, disability, MRI, safety, and antigenicity. Neurological assessments were conducted every 3 months. The primary endpoint of the study was mean relapse count per patient at 2 years.1
PRISMS TRIAL: INITIATED IN 19941
Relapses
REBIF® DEMONSTRATED SIGNIFICANT REDUCTIONS IN RELAPSES IN PATIENTS WITH A RANGE OF DISABILITY1-3
Demonstrated reduction in the mean number of relapses vs placebo at 2 years1,2


Fewer patients had a relapse at 3 months with Rebif® vs placebo3
TOTAL PATIENT COHORT (BASELINE EDSS 0 TO 5.0)


SC: subcutaneous; tiw: 3 times weekly.
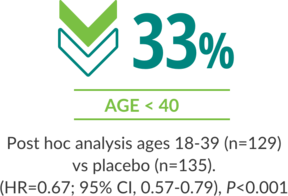
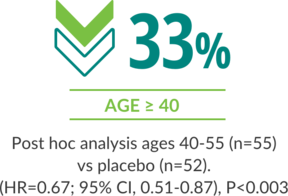
Regardless of age, Rebif® demonstrated a reduction in ARR at 2 years1,4,5
ARR AT 2 YEARS IN AGES 18-55 VS PLACEBO (BASELINE EDSS 0 TO 5.0)
ARR: annualized relapse rate; CI: confidence interval; EDSS: Expanded Disability Status Scale; HR: hazard ratio.

Regardless of gender, Rebif® demonstrated a reduction in relapse at 2 years2


MRI RESULTS
REBIF® DEMONSTRATED A SIGNIFICANT REDUCTION IN MRI LESIONS1-3
Patients on Rebif® showed a 78% reduction in T2-active lesions vs placebo after 2 years1,2
Median number of T2-active lesions per patient per scan was 0.5 for 44 mcg (n=171) vs 2.25 for placebo (n=172); P<0.0001*

Significant CUA lesion reduction seen as early as 2 months and persisted up to 9 months2†‡§
MONTHLY SCANS—MEAN NUMBER OF CUA LESIONS OVER TIME

*Based on comparisons from rank-based ANOVA.2
†From a subgroup of 205 patients who underwent monthly MRI scans for the first 9 months, intent-to-treat population had biannual scans.6
‡Analysis of the subgroup of patients undergoing PD/T2 and T1-Gd+ MRI scans at prestudy day 1, and monthly for the first 9 months of treatment based on ANOVA on the ranks taking center and number of active lesions at baseline into account.2
§Proportion of scans per patient per treatment group with new activity, including new, enlarging, recurrent PD/T2, or enhancing T1 lesions.6
CUA: combined unique active, defined as any lesion that was T1 active or T2 active.
SC: subcutaneous; tiw: 3 times weekly.
Post hoc analysis of a 205-patient subset who received monthly MRI scans in the PRISMS study.

PD/T2 lesion load also decreased significantly compared with placebo1,6
DECREASED PD/T2 LESION LOAD: BIANNUAL SCANS6

N values: At 6 months, placebo (n=182), Rebif® 22 mcg (n=182), Rebif® 44 mcg (n=182); at 12 months, placebo (n=179), Rebif® 22 mcg (n=180), Rebif® 44 mcg (n=180); at 18 months, placebo (n=176), Rebif® 22 mcg (n=177), Rebif® 44 mcg (n=172); and at 24 months, placebo (n=172), Rebif® 22 mcg (n=171), Rebif® 44 mcg (n=171).6 Lesion load is defined as the total area of lesions in the brain measured in mm2.6
PD: proton density; SC: subcutaneously; tiw: 3 times weekly.
- Both Rebif® groups exhibited a decrease in T2 lesion load, which was evident by 6 months of observation; at 2 years, there was a 1.2% and 3.8% reduction in the 22 mcg and 44 mcg groups, respectively (P<0.0001 compared with placebo for each group)
- The rate of serious adverse events in patients with EDSS >3.5 to 5.0 (regardless of causality) was 20% versus 11.3% for the total patient cohort (EDSS 0 to 5.0)

T1-GD+ lesions showed a significant reduction compared with placebo6
SIGNIFICANT REDUCTIONS IN T1-GD+ LESIONS AT 9 MONTHSII¶

IIFrom a subgroup of 205 patients who underwent monthly MRI scans for the first 9 months, intent-to-treat population had biannual scans.
¶Analysis of the subgroup of patients undergoing PD/T2 and T1-Gd+ MRI scans at prestudy screening, study day 1, and monthly for the first 9 months of treatment based on ANOVA on the ranks taking center and number of active lesions at baseline into account.
SC: subcutaneous; tiw: 3 times weekly; T1-Gd+: T1-weighted gadolinium-enhancing.
Disability progression
REBIF® DEMONSTRATED A SIGNIFICANT DELAY IN DISABILITY PROGRESSION VS PLACEBO IN PATIENTS WITH A RANGE OF DISABILITY1,2
TOTAL PATIENT COHORT (BASELINE EDSS 0 TO 5.0)

#Progression of disability was defined as an increase of at least 1 point in the EDSS, sustained for at least 3 months.
CDP: confirmed disability progression; HR: hazard ratio; SC: subcutaneous; tiw: 3 times weekly.

HIGHER EDSS COHORT (BASELINE EDSS >3.5 TO 5.0)

**Progression of disability was defined as an increase of at least 1 point in the EDSS, sustained for at least 3 months.
CDP: confirmed disability progression; HR: hazard ratio; SC: subcutaneous; tiw: 3 times weekly.
- In time to confirmed disability progression, a statistically significant difference was seen between
Rebif® 22 mcg and placebo in the total patient cohort (0 to 5.0) but was not seen in the high EDSS cohort (>3.5 to 5.0) - The rate of serious adverse events in patients with EDSS >3.5 to 5.0 (regardless of causality) was 20% vs 11.3% for the total patient cohort (EDSS 0 to 5.0)
INDICATION AND IMPORTANT SAFETY INFORMATION
for REBIF® (interferon beta-1a) for subcutaneous injection
INDICATION
Rebif is indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
IMPORTANT SAFETY INFORMATION
Contraindication: Rebif is contraindicated in patients with a history of hypersensitivity to natural or recombinant interferon beta, human albumin, or any other component of the formulation.
Depression and Suicide: Use Rebif with caution in patients with depression, a common condition in people with multiple sclerosis. Depression, suicidal ideation, and suicide attempts have been reported to occur with increased frequency in patients receiving interferon compounds, including Rebif.
Hepatic Injury: There have been rare reports of severe liver injury, including some cases of hepatic failure requiring liver transplantation, in patients taking Rebif. Consider the potential for hepatic injury when Rebif is used in combination with other products associated with hepatotoxicity. Monitor liver function tests and patients for signs and symptoms of hepatic injury. Consider discontinuing Rebif if hepatic injury occurs.
Anaphylaxis and Other Allergic Reactions: Anaphylaxis and other allergic reactions (some severe) have been reported. Discontinue Rebif if anaphylaxis occurs.
Injection Site Reactions Including Necrosis: In controlled clinical trials, injection site reactions occurred more frequently in Rebif-treated patients than in placebo-treated and Avonex-treated patients. Injection site reactions including injection site pain, erythema, edema, cellulitis, abscess, and necrosis have been reported in the postmarketing setting with the use of Rebif. Do not administer Rebif into affected area until fully healed; if multiple lesions occur, change injection site or discontinue Rebif until skin lesions are healed. Some cases of injection site necrosis required treatment with intravenous antibiotics and surgical intervention (debridement and skin grafting). Some cases of injection site abscesses and cellulitis required treatment with hospitalization for surgical drainage and intravenous antibiotics. Rotate site of injection with each dose to minimize likelihood of severe injection site reactions, including necrosis or localized infection.
Decreased Peripheral Blood Counts: Decreased peripheral blood counts in all cell lines, including pancytopenia, have been reported in Rebif-treated patients. In controlled clinical trials, leukopenia occurred at a higher frequency in Rebif-treated patients than in placebo and Avonex-treated patients. Thrombocytopenia and anemia occurred more frequently in 44 mcg Rebif-treated patients than in placebo-treated patients. Monitor patients for symptoms or signs of decreased blood counts. Monitoring of complete blood and differential white blood cell counts is also recommended.
Thrombotic Microangiopathy: Cases of thrombotic microangiopathy (TMA), some fatal, have been reported with interferon beta products, including Rebif, up to several weeks or years after starting therapy. Discontinue Rebif if clinical symptoms and laboratory findings consistent with TMA occur and manage as clinically indicated.
Pulmonary Arterial Hypertension: Cases of pulmonary arterial hypertension (PAH) have been reported in patients treated with interferon beta products, including REBIF. Patients who develop unexplained symptoms (e.g., dyspnea, new or increasing fatigue) should be assessed for PAH. If alternative etiologies have been ruled out and a diagnosis of PAH is confirmed, discontinue treatment and manage as clinically indicated.
Seizures: Seizures have been temporally associated with the use of beta interferons, including Rebif, in clinical trials and in postmarketing reports. Monitor for seizures when administering Rebif to patients, particularly those with pre-existing seizure disorders.
Laboratory Tests: New or worsening thyroid abnormalities have developed in some patients treated with Rebif. Thyroid function tests are recommended every 6 months in patients with history of thyroid dysfunction or as clinically indicated.
Adverse Reactions: The most common side effects with Rebif are injection-site disorders, influenza-like symptoms, abdominal pain, depression, elevated liver enzymes, and hematologic abnormalities.
Pregnancy: Epidemiological data do not suggest a clear relationship between interferon beta use and major congenital malformations, but interferon beta may cause fetal harm based on animal studies. Data from a large human population-based cohort study, as well as other published studies over several decades, have not identified an increased risk of major birth defects with exposure to interferon beta products during early pregnancy. Findings regarding a potential risk for low birth weight or miscarriage with the use of interferon beta products in pregnancy have been inconsistent.
Lactation: Limited published literature has described the presence of interferon beta-1a products in human milk at low levels. There are no data on the effects of interferon beta-1a on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for REBIF and any potential adverse effects on the breastfed child from REBIF or from the underlying maternal condition.
To report SUSPECTED ADVERSE REACTIONS, contact EMD Serono, Inc. at 1-800-283-8088 ext. 5563 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Please see Full Prescribing Information and Medication Guide.
References: 1. PRISMS Study Group. Randomised double-blind placebo-controlled study of interferon β-1a in relapsing/remitting multiple sclerosis. Lancet. 1998;352(9139):1498-1504. 2. Data on file. EMD Serono, Inc. PRISMS study report. 3. Data on file. EMD Serono, Inc. PRISMS 2010 Confirming analysis. 4. Rebif® [Prescribing Information]. Rockland, MA: EMD Serono, Inc. 5. Freedman MS, Brod S, Wray S, et al. Post hoc Analysis to Evaluate the Effects of Subcutaneous Interferon beta-1a in Subgroups of Patients from the PRISMS Study with Early Onset Versus Late Onset Disease. Poster presented at: ECTRIMS 2019; 11-13 September; Stockholm, Sweden. 6. Li DKB, Paty DW; UBC MS/MRI Analysis Research Group, PRISMS Study Group. Magnetic resonance imaging results of the PRISMS trial: a randomized double-blind, placebo-controlled study of interferon-β1a in relapsing-remitting multiple sclerosis. Ann Neurol. 1999;46(2):197-206.
INDICATION AND IMPORTANT SAFETY INFORMATION
for REBIF® (interferon beta-1a) for subcutaneous injection
INDICATION
Rebif is indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
IMPORTANT SAFETY INFORMATION
Contraindication: Rebif is contraindicated in patients with a history of hypersensitivity to natural or recombinant interferon beta, human albumin, or any other component of the formulation.
Depression and Suicide: Use Rebif with caution in patients with depression, a common condition in people with multiple sclerosis. Depression, suicidal ideation, and suicide attempts have been reported to occur with increased frequency in patients receiving interferon compounds, including Rebif.
Hepatic Injury: There have been rare reports of severe liver injury, including some cases of hepatic failure requiring liver transplantation, in patients taking Rebif. Consider the potential for hepatic injury when Rebif is used in combination with other products associated with hepatotoxicity. Monitor liver function tests and patients for signs and symptoms of hepatic injury. Consider discontinuing Rebif if hepatic injury occurs.
Anaphylaxis and Other Allergic Reactions: Anaphylaxis and other allergic reactions (some severe) have been reported. Discontinue Rebif if anaphylaxis occurs.
Injection Site Reactions Including Necrosis: In controlled clinical trials, injection site reactions occurred more frequently in Rebif-treated patients than in placebo-treated and Avonex-treated patients. Injection site reactions including injection site pain, erythema, edema, cellulitis, abscess, and necrosis have been reported in the postmarketing setting with the use of Rebif. Do not administer Rebif into affected area until fully healed; if multiple lesions occur, change injection site or discontinue Rebif until skin lesions are healed. Some cases of injection site necrosis required treatment with intravenous antibiotics and surgical intervention (debridement and skin grafting). Some cases of injection site abscesses and cellulitis required treatment with hospitalization for surgical drainage and intravenous antibiotics. Rotate site of injection with each dose to minimize likelihood of severe injection site reactions, including necrosis or localized infection.
Decreased Peripheral Blood Counts: Decreased peripheral blood counts in all cell lines, including pancytopenia, have been reported in Rebif-treated patients. In controlled clinical trials, leukopenia occurred at a higher frequency in Rebif-treated patients than in placebo and Avonex-treated patients. Thrombocytopenia and anemia occurred more frequently in 44 mcg Rebif-treated patients than in placebo-treated patients. Monitor patients for symptoms or signs of decreased blood counts. Monitoring of complete blood and differential white blood cell counts is also recommended.
Thrombotic Microangiopathy: Cases of thrombotic microangiopathy (TMA), some fatal, have been reported with interferon beta products, including Rebif, up to several weeks or years after starting therapy. Discontinue Rebif if clinical symptoms and laboratory findings consistent with TMA occur and manage as clinically indicated.
Pulmonary Arterial Hypertension: Cases of pulmonary arterial hypertension (PAH) have been reported in patients treated with interferon beta products, including REBIF. Patients who develop unexplained symptoms (e.g., dyspnea, new or increasing fatigue) should be assessed for PAH. If alternative etiologies have been ruled out and a diagnosis of PAH is confirmed, discontinue treatment and manage as clinically indicated.
Seizures: Seizures have been temporally associated with the use of beta interferons, including Rebif, in clinical trials and in postmarketing reports. Monitor for seizures when administering Rebif to patients, particularly those with pre-existing seizure disorders.
Laboratory Tests: New or worsening thyroid abnormalities have developed in some patients treated with Rebif. Thyroid function tests are recommended every 6 months in patients with history of thyroid dysfunction or as clinically indicated.
Adverse Reactions: The most common side effects with Rebif are injection-site disorders, influenza-like symptoms, abdominal pain, depression, elevated liver enzymes, and hematologic abnormalities.
Pregnancy: Epidemiological data do not suggest a clear relationship between interferon beta use and major congenital malformations, but interferon beta may cause fetal harm based on animal studies. Data from a large human population-based cohort study, as well as other published studies over several decades, have not identified an increased risk of major birth defects with exposure to interferon beta products during early pregnancy. Findings regarding a potential risk for low birth weight or miscarriage with the use of interferon beta products in pregnancy have been inconsistent.
Lactation: Limited published literature has described the presence of interferon beta-1a products in human milk at low levels. There are no data on the effects of interferon beta-1a on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for REBIF and any potential adverse effects on the breastfed child from REBIF or from the underlying maternal condition.
To report SUSPECTED ADVERSE REACTIONS, contact EMD Serono, Inc. at 1-800-283-8088 ext. 5563 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Please see Full Prescribing Information and Medication Guide.
US-REB-00280
Last update 04/2024
This website is intended for U.S. healthcare professionals only.
Use and access of this site are subject to the terms and conditions set out in our Legal Statement and Privacy Policy.
EMD Serono is the Healthcare business of Merck KGaA, Darmstadt, Germany in the U.S. and Canada. Rebif, MS LifeLines, Rebiject II, Rebidose, and the Rebif logo are registered trademarks of Merck KGaA, Darmstadt, Germany, or its affiliates.
©2024 Merck KGaA, Darmstadt, Germany or its affiliates. All rights reserved. EMD Serono Inc.
US-REB-00280
Last update 04/2024
©2024 Merck KGaA, Darmstadt, Germany or its affiliates. All rights reserved.